If you’ve ever had to get figures ready for a paper, present data to your group, or even just process a large amount of NMR data, you’ve probably found a lot of NMR data processing to be… repetitive. Luckily, you’re not the only one! Automating the small tasks - like FT, or zero-filling, or peak picking a specific region in your data could take minutes to set up and save you hours of work. As an added bonus, if you hard code these in as Macros, your processing will be specific for your data and consistent across all data you’re working with.
So, the first question when processing is, “Can I automate this”. For a good 80% of processing, absolutely. Many labs have their own Macros set up for automatic FT, phase correction, and NUS reconstruction for 2D+ data - but this all is specific to who your facilities manager is, what tasks they saw fit to automate, and so on. If your looking at a pile of metabolomics 1D data, you’d be hard pressed to find a justification for doing these basic commands individually.
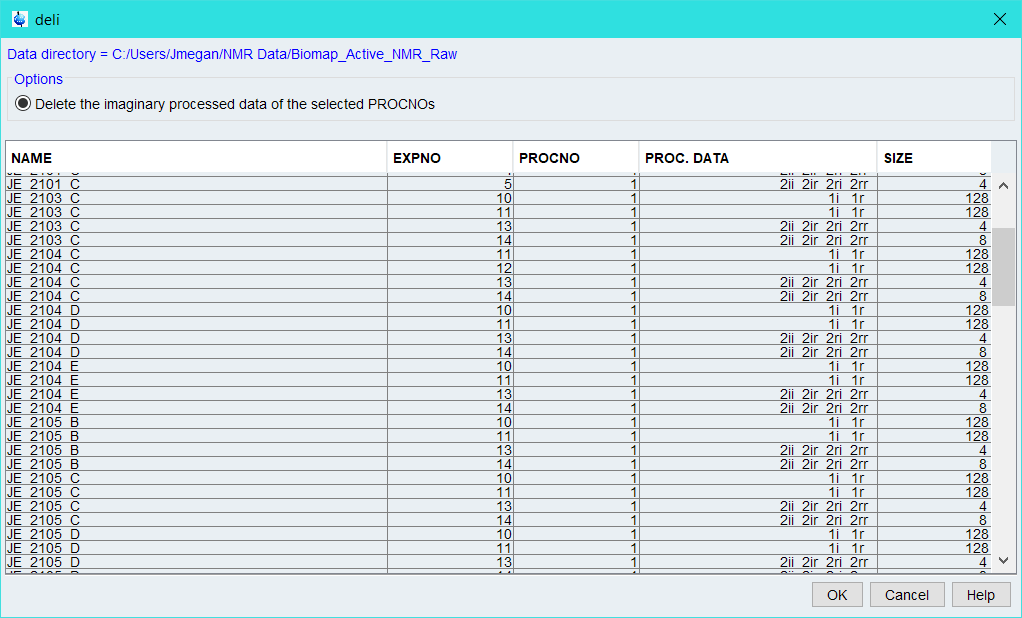
Before we jump in - there is a limit. I found this limit, repeatedly, when I was attempting to peak pick metabolomics 2D data. If you asked me if it could be done, I’d say yes - but I’d also say that it involves a lot more than what you may consider. For that reason, I have largely abandoned this single task in my automation pipelines and have opted for hand picking my data. However, if you’re working with pure molecules in a reasonable concentration, this is a much easier task. When I was originally looking into ways to make this a feasible task, a discussion between Clemens Anklin and John Hollerton put forth the idea of 80:20 confidence - that for around 80% of cases of automation, it’s possible to get great results, the other 20%, however, might prove challenging. For this reason, you should always manually check your data after an automated pipeline. In fact… you could even automate this…. but I digress.
Over the next few posts, I hope to shed some light on how to automate your NMR processing using Topspin. There exists some automation documentation in the Topspin Manual, but for the average user, the information can be overwhelming and it can take days of working through it to automate your first task. This is NOT a criticism! They are highly detailed and contain a lot of information that might be of interest. After these small blog posts, my hope is that you’ll be able to get started with automation in under 30 minutes and then progress to the manuals to fill in the gaps - once you know what you’re looking for, it’s easier to find.
Types of Automation
Topspin contains (at least) 3 different ways to automate your processing - and it’s accessible to all users, even on an academic license.
They are:
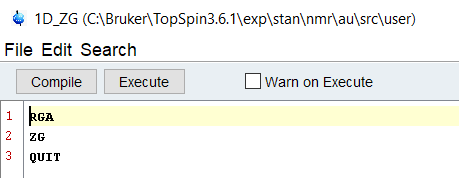
For the average user who knows nothing of coding, the simplest way to get started is by using Macros. I say this because when you program a macro in Topspin, you simply replace the human with a series of commands - if you already type things into the command line in topspin, you already know how to do it.
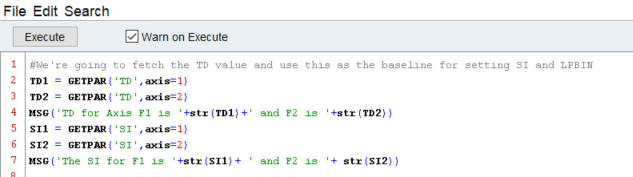
Other users, such as myself, who are familiar with Python will rejoice to learn that the python scripting in topspin can be used to read/write files - such as peak lists - or perform calculations on data and export the results. This can save you a massive amount of time in ensuring you have the right peak lists saved with the right file names in the right places, and if you already know basic python, Topspin’s python is really easy to use.
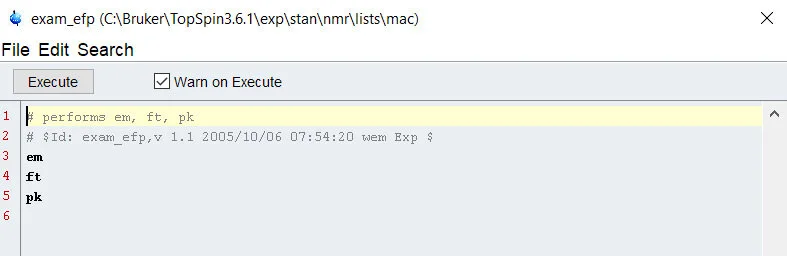
The last type, Au Scripts, are higher level programming scripts that must be compiled before execution. The code used recognizes topspin commands and C-language functions. The benefit, of course, is that it is extremely adaptable and very fast when compared head to head with python and it carries the same ability to export data easily. However, if you’ve not used a C based language, it can take some getting used to.
There are other factors that play into what to decide to automate with, but I find that people use the tools they’re already familiar with - we all have a toolbox filled with screwdrivers, but we all have a favorite (mine happens to be an old Craftsman given to me by my grandfather). MestreNova has it’s own scripting ability, but I will not be going into this toolchest - it’s someone else’s set.
I hope to create a post every Tuesday for the next 3 weeks to highlight each of these options, how to use them, and some simple scripts to get you started.